


固相法合成螺环磷酰哌嗪酯阻燃剂
摘要:以螺环磷酰二氯(SPDPC)与哌嗪为原料,在未加溶剂和催化剂的条件下,用高速万能粉碎机混合原料的同时,借助转刀高速旋转的剪切刀所产生的热能加热体系发生反应,合成了螺环磷酰哌嗪酯。分析了原料的摩尔比以及混合时间对产品得率的影响,得出了较优的反应条件:n(螺环磷酰哌嗪酯:)n(哌嗪)=1 : 2,混合时间350s ,在该条件下得到暗白色粉末状固体,产率达95%以上。产品用红外光谱和核磁共振波谱进行了表征。
关键词:螺环磷酰二氯;哌嗪;固相反应;阻燃剂;橡塑助剂
磷氮阻燃剂是一类性能优良的膨胀阻燃剂。适用于天然纤维及合成高分子材料,具有低烟、无毒等优点。含氮的螺环磷酸酯属于膨胀阻燃体系,对聚丙烯有良好的阻燃性能且对聚丙烯的力学性能影响较小。以螺环磷酰二氯(SPDPC)与哌嗪为原料乙腈作溶剂,三乙胺作催化剂,首次合成了初始分解温度及成炭率都较高的膨胀型阻燃剂螺环磷酰哌嗪酯。
本文利用高速超细粉碎机,将螺环磷酰二氯(SPDPC)与哌嗪按一定摩尔比混合,在没有溶剂和催化剂的存在下,在粉碎混合的同时,利用转刀高速旋转所产生的热能,使体系加热,发生反应,合成了螺环磷酰哌嗪酯。方法绿色环保,简单易行。固相法合成阻燃剂,可以免去反应介质溶剂的使用,不但降低合成成本,而且减少有机溶剂的挥发,保护环境,符合低碳化工的要求。
1 实验部分
1.1 试剂与仪器
FW—80高速万能粉碎机;Magna—IR 560 esp型傅里叶变红外光谱仪,;德国Beuker Advance Ⅲ400型核磁共振波谱仪,氚代三氟乙酸作溶剂,DMS作内标;三氯氧磷、季戊四醇和无水哌嗪均为AR 。
1.2 产品合成
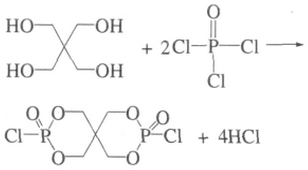
1.2.1 SPDPC的合成
合成SPDPC。反应式如下:
1.2.2 螺环磷酰哌嗪酯的固相合成
将SPDPC与哌嗪按一定摩尔比混合,在高速万能粉碎机中,粉碎一定时间。取出,冷却,用冰的二甲基甲酰胺(DMF)洗涤,得到暗白色粉末状固体。原料摩尔比及反应时间设计如表1所示。反应式如下: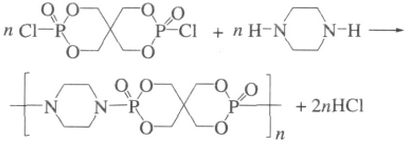
1.3 螺环磷酰哌嗪酯的结构表征
1.3.1 红外光谱测定
取较优条件下合成的产品,在Magna—IR 560 esp型傅里叶变红外光谱仪上测定其红外光谱图。
1.3.2 NMR测定
取较优条件下合成的产品,在德国德国Beuker Advance Ⅲ400型核磁共振波谱仪(氚代三氟乙酸作溶剂)上测定其111NMR谱、13CNMR以及31PNMR谱图。
2 结果与讨论
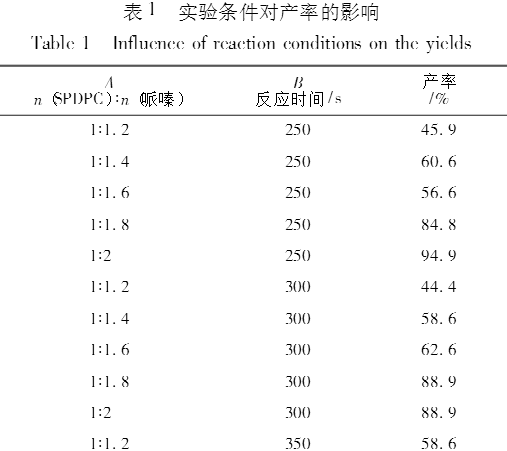
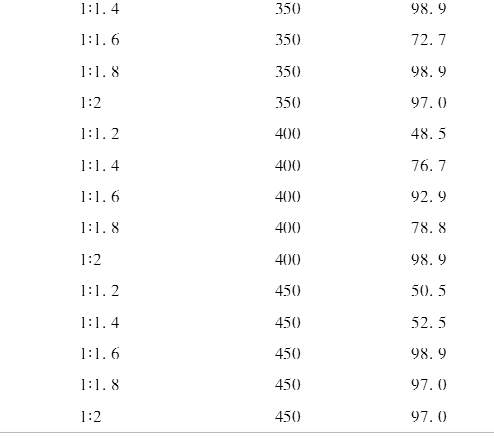
2.1 螺环磷酸哌嗪酯最佳合成条件的确定
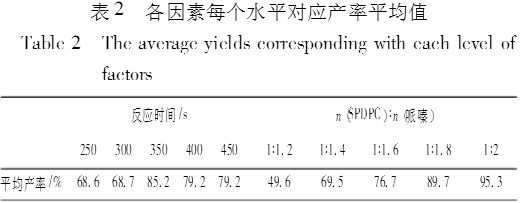
实验结果如表1所示、可以看出,产率与反应时间和原料摩尔比都相关。把各因素对应产率的平均值列在表2中。从表2可以看出,反应时间为350s时,平均产率最高,时间再延长,平均产率反而下降,所以最佳反应时间是350s。从原料摩尔比看,随着哌嗪在混合物中的比例增加,平均产率逐渐增加。中间体SPDPC与哌嗪摩尔比为1:2时,平均产率达到95.3%。如果继续增加哌嗪的量,平均产率也许还会增加,但是空间已经不大。所以本实验选取的最佳原料摩尔比为1:2 。因此,最佳合成条件确定为中间体SPDPC与哌嗪以1:2摩尔比混合,反应350s 。
2.2 产品的波谱表征及其聚合度测定
2.2.1 红外光谱
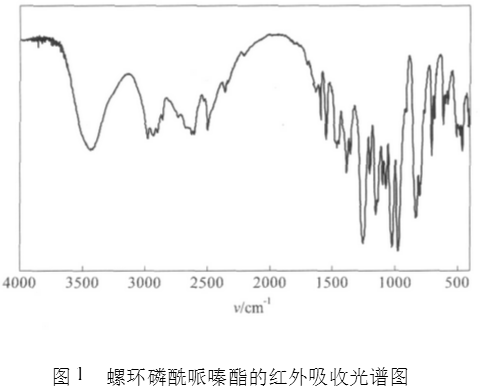
最佳条件下合成螺环磷酰哌嗪酯的红外光谱图如图1所示。图1中P=O的吸收峰为1 255cm-1 , P-N-C吸收峰为968cm-1,P-O-C的吸收峰为1 100~1 000cm-1,螺环吸收峰为910、823、799及682cm-1。
2.2.2 NMR核磁共振波谱
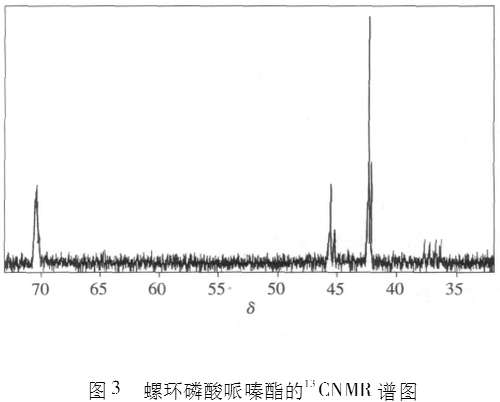
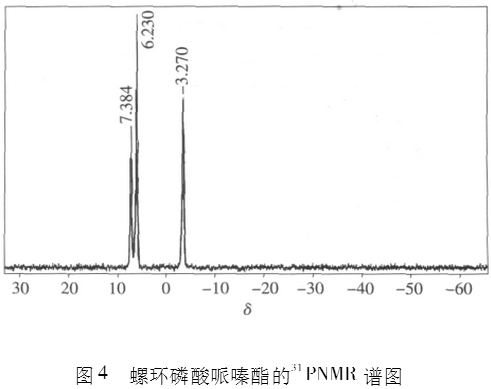
图2为螺环磷酸哌嗪酯的1IINMR谱,图3为螺环磷酸哌嗪酯的13CNMR谱,图4为螺环磷酸哌嗪酯的31PNMR谱。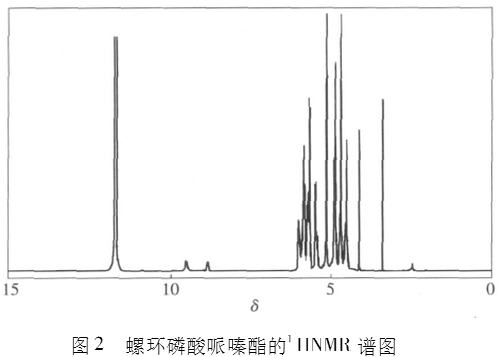
从图2螺环磷酰哌嗪酯的1IINMR谱中可以看出,螺环II分别在5.8、5.5以及5.1处产生3组峰,这3组峰总积分面积42+35+45=122;在4.8处出现了哌嗪环上的II峰,总积分面积为86,螺环II与哌嗪环II个数比122:86≈3:2。
从图3螺环磷酸哌嗪酯的13CNMR谱上可以看出,42.2和40.5处为螺环上5个C的信号,总积分面积(2.00+0.99)=3,70.3处为哌嗪环上C的信号,积分面积1.52。螺环上的C与哌嗪环上的C的积分面积之比约2 : 1 。
从图4螺环磷酸哌嗪酯的31PNMR谱可以看出,7.384和6.230处是P-N中P的吸收峰,积分面积1.34;二者比例约为2:1。相当于分子中有2个P-N键,同时会有1个P-CI键。可能的结构应该是3个螺环与2个哌嗪聚合,两端是P-CI。那么螺环磷酰哌嗪酯的结构可能如图5所示。此时螺环上的C数(3×5=15)与哌嗪环上的C数(2×4=8)约为2:1。与13CNMR谱推测的结果一致。此时,螺环上的II原子与哌嗪环上的II原子个数之比约为3:2,与1IINMR谱推测的结果一致。
公司经营产品:
